Metastatic melanoma used to have a very poor prognosis, which has improved since the introduction of immunotherapy (immune checkpoint inhibition; ICI) and targeted therapy. Although some patients attain long-term disease control and survival with ICI, about 50% of patients do not respond. As the median time to response is 2-5 months, considering the possibility of late responses and pseudo-progression, most patients have been treated for over 5 months before being identified as a non-responder, putting them at risk for severe toxicity. Furthermore, for nonresponders, alternative treatment could have been initiated earlier, possibly with better response changes. Lastly, ICI are expensive, with ~50,000 Euro per patient for 5 months treatment. Therefore, a reliable and affordable personalized response prediction algorithm could reduce costs, optimize treatment efficacy and reduce unnecessary side effects..
In the PREMIUM project we aim to predict non-response to ICI by applying machine learning techniques to clinical and genomic data and pathology and CT scan images from 1500 immunotherapy treated melanoma patients. These data and images are routinely acquired during standard of care for every patient. A joint neural network based on both types of data (clinicopathological data and histological and CT images) is developed and trained to evaluate the predictive performance of combining these different data sources.
Immunotherapy with Immune Checkpoint Inhibitors (ICI) has been a major breakthrough in cancer treatment. However, despite its promise of being able to cure patients from metastatic cancer, many patients do not respond to ICI. Moreover, while being well-tolerated by some patients, ICI cause severe and even fatal immune-related toxicity in others. Large cohorts structurally analyzing immune subsets in tissue and circulating cells are needed for:
- Upfront identification of non-responders
- Identifying leads for alternative (immuno)therapies
- Evidence based toxicity management to effectively treat ICI toxicity without compromising its anti-cancer effect
The UNICIT biobank is a unique biobank cohort containing data, blood, feces and tissue from ICI treated patients (see figure). Having included longitudinal blood and tissue samples from over 500 patients with ongoing recruitment, this biobank is the optimal source for studying ICI resistance and toxicity using AI approaches.

Some examples:
- Local immune cell composition and function in tissue (Nanostring, scRNAseq, imaging mass cytometry)
- Peripheral immune responses in blood before and after treatment (Olink/Luminex, flow cytometry)
- Microbiome composition (16S rRNAseq, Metagenomic shotgun sequencing)
About 97% of all mutations in cancer occur in non-coding regions of the genome. These regions are packed with cis-regulatory elements (CREs) such as promoters, enhancers. Non-coding mutations within these CREs can affect gene expression of cancer-relevant genes, thereby classifying them as non-coding driver mutations. Although anecdotal examples exist for these driver mutations, global, genome-wide statistical based methods aimed at identifying these driver mutations have led to a limited number of discoveries. This is partially because non-coding mutations are inherently more difficult to interpret than coding mutations. The goal of the PERICODE consortium (consisting of labs from NKI, UMCU, Groningen and A-UMC) is to develop a computational algorithm that can predict the impact of non-coding mutations from DNA sequence alone.
Towards this goal, we combine a functional high-throughput assay with a residual neural network to predict and study the effects of non-coding cancer mutations on gene-expression. We use a massively parallel reporter assay (MPRA) in which millions of unique DNA fragments of ~300 bp can be tested for their promoter activity. Remarkably, the neural network trained on these MPRA data is able to accurately predict promoter activity. As our model is able to predict promoter activity from any sequence up to 600 bp, it can make predictions of the effects of any non-coding mutation in the genome on local promoter activity. Initial benchmarking the CNN with available mutagenesis data indicates that our model can indeed predict such effects quite well (R in the range of 0.5). We are implementing an integrated wet-lab / dry-lab strategy to further improve and rigorously characterize this performance, and to apply the methodology to a broad diversity of tumor types. This should provide fundamental insights into the overall impact of non-coding mutations in cancer, and enable the discovery of novel variants relevant for specific cancers.
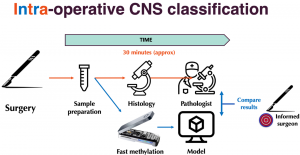
Molecular classification of tumor subtypes is essential for optimal treatment. For central nervous system (CNS) tumors, for instance, it is clear that tumor subtype should determine surgical strategy. However due to a lack of pre-operative tissue-based diagnostics, limited knowledge of the precise tumor type is available at the time of surgery. Using real-time nanopore sequencing, a sparse methylation profile can be directly obtained during surgery, making it ideally suited to enable intraoperative diagnostics.
The de Ridder lab at the Center for Molecular Medicine of the UMC Utrecht, in collaboration with the Princess Máxima Center for pediatric oncology, developed a state-of-the-art deep learning approach called Sturgeon, to deliver trained models that are lightweight and universally applicable across patients and sequencing depths. The method is highly accurate and fast enough to provide a correct diagnosis with as little as 20 to 40 minutes of sequencing data in 45 out of 49 pediatric samples, and inconclusive results in the other four. In four intraoperative cases we achieved a turnaround time of 60-90 minutes from sample biopsy to result; well in time to impact surgical decision making. This work demonstrates that machine-learned diagnosis based on intraoperative sequencing can assist neurosurgical decision making, allowing neurological comorbidity to be avoided or preventing additional surgeries.